Isotopologue Library¶
-
class
pyqms.IsotopologueLibrary(molecules=None, charges=None, metabolic_labels=None, fixed_labels=None, params=None, trivial_names=None, verbose=True, evidences=None)¶ The Isotopologue library is the core of pyQms.
Keyword Arguments: - molecules (list of str) – Molecules used to build the library, for more details see below.
- charges (list of int) – Charge list used to build the library
- metabolic_labels (dict) – see below
- fixed_labels (dict) – see below
- params (dict) – Match parameters, see pyqms.params
- trivial_names (dict) – Dictionary that is used to build up lookups. Key is a molecule and value a trivial name.
- evidences (dict) – Dictionary that is used to build up additional lookups. Key is a formula pointing to a subdict. Subdict has molecules as keys and values are ‘trivial_names’ as a list and ‘evidences’ holding evidence/identification information
- verbose (bool) – Be verbose or not during initialization and matching.
Keyword argument examples:
molecules The molecule format can be anything that the ChemicalComposition class understands. Currently this can for example be:
[ '+{0}'.format('H2O'), '{peptide}'.format(peptide='PEPTIDE'), '{peptide}+{0}'.format('PO3', peptide='PEPTIDE'), '{peptide}#{unimod}:{pos}'.format( peptide = peptide, unimod = 'Oxidation', pos = 1 ) ]
metabolic_labels is used to define new element pools with enriched isotopes. The dict key defines an enriched element, e.g. 15N or 13C and its value is a list of floats [0 - 1.0] defining enrichment.The combination of those pools is used to calculate isotopologues:
{ '15N' : [0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 0.99] }
fixed_labels are based on unimod. Fixed molecules do not change the shape of the isotoplogue drastically but introduce a simple mass shift, like SILAC, 18O or others.
The format is for example:
{ 'R' : ['C(-6) 13C(6) N(-4) 15N(4)',''] }
Returns: Isotopologue library as dict where the top key is always the chemical forumla unimod style. Return type: dict simplified:
{ 'C(34)H(53)N(7)O(15)': { 'cc': { 'C': 34, 'H': 53, 'N': 7, 'O': 15 }, 'env': { (('N', '0.000'),): { # charge : 1: { # all transformed mz values 'atmzs': { 800443, 800444, # ... skipped 803459, 803460, 803461 }, # theoretical mz values 'mz': [ 800.4472772254203, 801.450389542063, 802.4536114854914, 803.4568170275203, 804.4597382487398, 805.463171867346, 806.4631454917885, 807.4676949759603 ], # transformed mz values within error # packages are on on peak level 'tmzs': [ { 800443, 800444, # ... skipped 800451 }, { 801446, 801447, # ... skipped 801454 }, { 802450, 802451, # ... skipped 802458 }, { 803453, 803454, 803455, # ... skipped 803461 }, None, None, None, None ] }, # charge independent information 'abun': [ 64799, 26251, 7164, 1456, 175, 20, 1, 0 ], 'c_peak_pos': [ 0, 1, 2, 3, None, None, None, None ], 'isot': [], 'mass': [ 799.3599640346001, 800.3629760500413, 801.3660976813065, 802.3692029128123, 803.3720238519379, 804.3753571372156, 805.3752307742944, 806.3796798135622 ], 'n_c_peaks': 4.0, 'relabun': [ 1.0, 0.40511743373159037, 0.11054965400744385, 0.022466784140529883, 0.002693331560888158, 0.0003019650321460501, 7.716705830708012e-06, 3.639837831552297e-08 ] } } } }
-
match_all(mz_i_list=None, file_name=None, spec_id=None, spec_rt=None, results=None)¶ Matches all isotopologues in the library agains a given mz_i_list
Parameters: - mz_i_list (list of tuples) – Spectrum information that should be matched against. Tuples of m/z and intensity
- file_name (str) – Information used for storage purpose. Useful if multiple files are parsed with one pyqms.result instance.
- spec_id (int) – Information used for storage purpose.
- spec_rt (float) – Information used for storage purpose.
- results (pyqms.Results) – (optional)
If a results object is passed to match_all, then this object will be updated and returned. This is for e.g. to accumulate results for a whole LC-MS/MS run.
For various examples using match_all please refer to the example scripts.
Returns: Object holding all quantitative information Return type: results class object (obj)
-
match_isotopologue(index=None, formula=None, charge=None, label_percentile=None, spec_tmz_set=None, spec_tmz_lookup=None, mz_i_list=None, mz_score_percentile=None)¶ Matches a single isotopologue onto a mz_i_list or spec_tmz_set
Parameters: - index (int) – Using this index one can retrieve all information about the molecule, i.e. lower_mz, upper_mz, charge, label_percentile, formula from self.formulas_sorted_by_mz. Alternatively, one can use the more verbose option: formula, charge and label_percentile
- formula (str) – pyqms formula type
- charge (int) – molecule charge
- label_percentile – pyQms label percentile
- mz_i_list (list of tuples) – List of m/z and intensity tuples, will be transformed to a spec_tmz_set given the defined precession. Alternatively, spec_tmz_set can be used as input.
- spec_tmz_set (set of ints) – tmz value set used for matching. Requires spec_tmz_lookup to get the actual mz which is required for scoring.
- mz_score_percentile (float) – Weighting of mz used for scoring. (1 - mz_score_percentile) is then intensity weighting. Values 0 - 1.0.
Note
Depending on the machine (some measure intensity better than others) adjusting mz_score_percentile value will give more accurate results. Best adjusted in pyqms.params (which can be passed during isotoplogue lib initialization)
Returns: Match results (tuple of score, scaling factor and matched peaks). - score reflects the fit of the theoretical isotopologue to the measured (both mz and intensities are compared)
- scaling factor reflects the actual amount of the molecule in the respective spectrum. It is defined as the sum of the total measured intensities divided by the sum of the total calculated intensities
- matched_peaks is list of tuples that contain measured_mz, measured_i, rel_i, calculated_mz, calculated_i
Multiple m/z values can occur in the range of the measured precision of every peak of the isotopologue, thus all combinations are considered and scored. Only the best scored match is returned for each isotopologue.
-
print_overview(formula, charge=None)¶ Prints an overview of a given molecule or formula to the std.out
Parameters: - formula (str) – Either formula or molecule
- charge (int) – Charge of the molecule
Examples
For PEPTIDE and charge 1:
Chemical formula C(34)H(53)N(7)O(15) (('N', '0.000'),) Isotope Abundance pos Mass m/z [MH]+1 transformed rel. 0 799.3599640346 800.4472772254 64799 1.00000000000 0 1 800.3629760500 801.4503895421 26251 0.40511743373 1 2 801.3660976813 802.4536114855 7164 0.11054965401 2 3 802.3692029128 803.4568170275 1456 0.02246678414 3 4 803.3720238519 804.4597382487 175 0.00269333156 None 5 804.3753571372 805.4631718673 20 0.00030196503 None 6 805.3752307743 806.4631454918 1 0.00000771671 None 7 806.3796798136 807.4676949760 0 0.00000003640 None
-
score_matches(matched_peaks, mz_score_percentile)¶ Score matched peaks.
Parameters: - matched_peaks (list of tuples) –
List of tuples containing
- measured_mz (mmz)
- measured_intensity (mi)
- relative_intensity_of_calculated_isotopologue_peak (ri)
- calculated_mz (cmz)
- calculated_i (ci)
- mz_score_percentile (float) – weighting of mz score
Parameters that influence the scoring are ‘MIN_REL_PEAK_INTENSITY_FOR_MATCHING’
Example plots
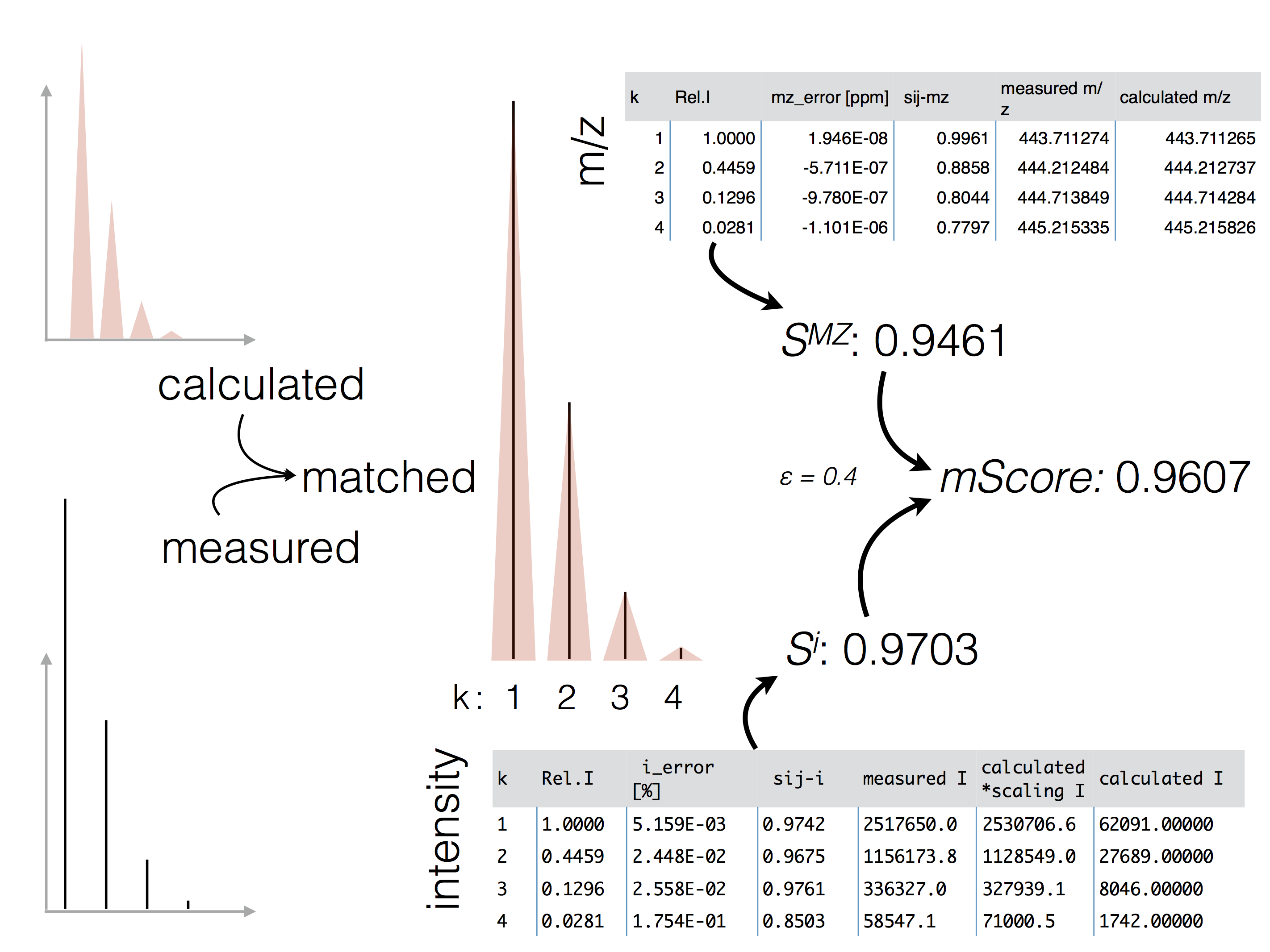
The figure below highlights the scoring principle. Erros for m/z and intensity values are determined and combined into the final mScore. For each peak of the isotopologue both errors are determined and influence the final score.
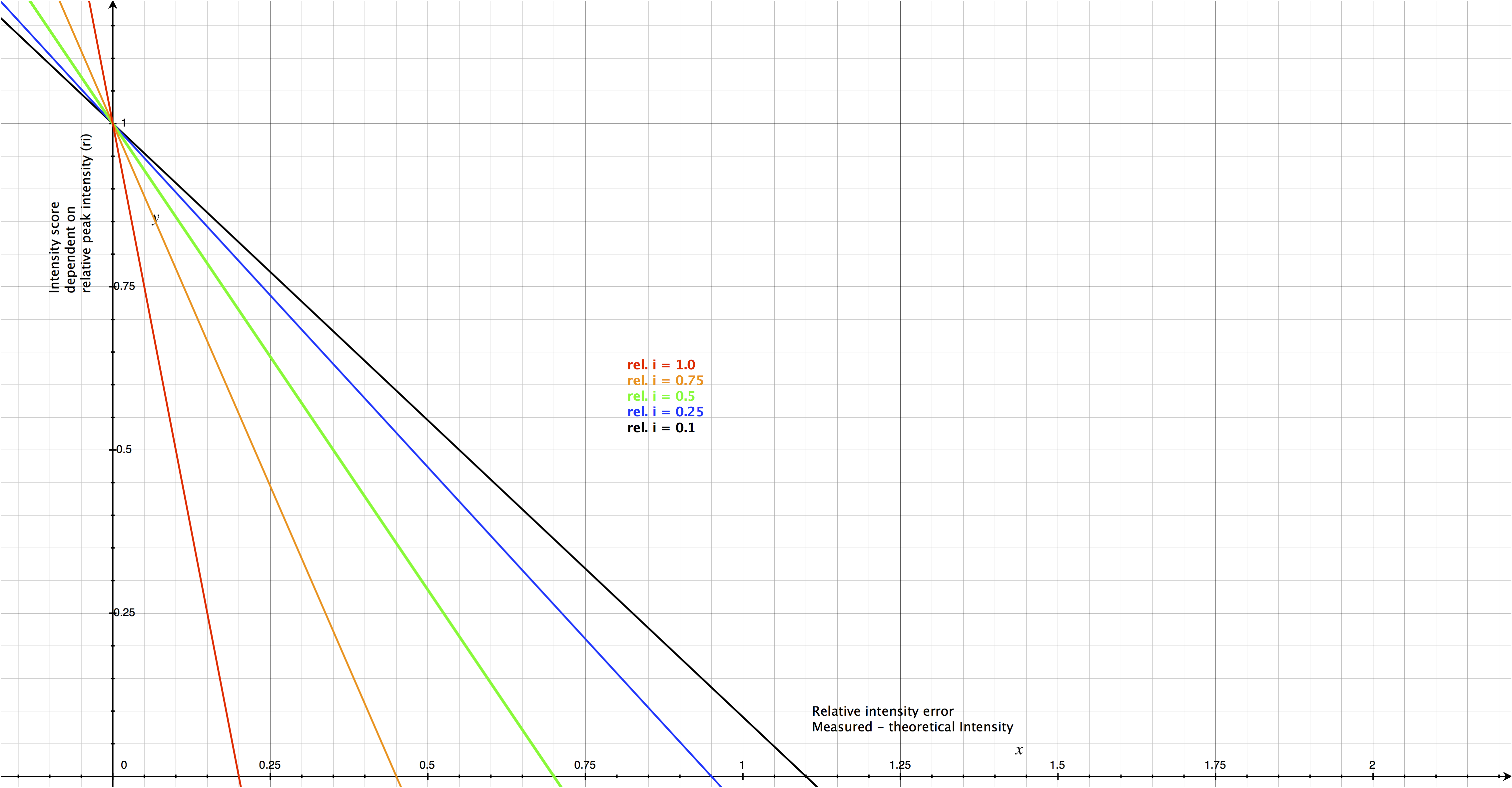
Calculated intensities are scaled to match the measured value and the deviation is calculated. The lower the intensity, the less accurate teh actual peaks are represented. To compensate for this, the intensity score decreases faster for large relative intensities compared to small relative intensities. This is highlighted in the following figure. Legend, x-axis represents the relative intensity error (measured - theoretical intensity) and the y-axis the intensity score. Different colors represent various relative peak intensities.
Scoring
Note
The proper display of the formulas of the next section requires access to the Internet when browsing the HTML documentation. The formulas are correctly embedded into the pdf of the documentation.
The pyQms matching score (mScore) is based on the work of Gower (1971) A General Coefficient of Similarity and Some of Its Properties, Biometrics (27), 857-871. The matching and scoring is performed on the m/z values and the intensity values independently yielding two scores, i.e. \(S^{mz}\) and \(S^{intensity}\). In both cases, each peak \(k\) is scored, comparing the measured value \(i\) with the calculated value \(j\) (equation 1), whereas a perfect match is 1. Each peak of the isotopologue that has a relative intensity (relative to the maximum intensity isotope peak) \(r_{k}\) above the matching threshold (by default 1% of the maximum intensity isotope peak) is matched and scored.
\begin{equation} s^{}_{ijk} \in [0, 1] \end{equation}The m/z score
For each peak \(k\), the m/z similarity between measured value \(i\) and the calculated value \(j\) is defined as
\begin{equation} s^{mz}_{ijk} = 1 - (\frac{\delta^{mz}_{ijk}}{\alpha}) \end{equation}Whereas \(delta^{mz}_{ijk}\) the difference in ppm between measured \(mz_{ik}\) and calculated \(mz_{jk}\) and \(\alpha\) defines the range in ppm, in which the score decreases from 1 to 0 in a linear fashion. In principle, \(\alpha\) is equal to the precision of the measurement defined by the user (pyQms parameter “REL_MZ_RANGE”, default 5 ppm, http://pyqms.readthedocs.io/en/latest/params.html). For example, if the difference between measured and theoretical m/z values would be 2.5 ppm, then the \(s^{mz}_{ijk}\) score for this peak \(k\) would be 0.5.
The total m/z score for all peaks termed \(S^{mz}\) is the weighted sum of all single similarity m/z scores \(s^{mz}_{ijk}\) (equation 3). The weighting is defined by the theoretical intensity of the peak \(k\) relative to the highest peak in the theoretical isotope pattern, termed \(r_{k}\).
\begin{equation} S^{mz} = \frac{\sum\limits_{}^k s^{mz}_{ijk} r_{k} }{\sum\limits_{}^k r_{k}} \end{equation}The intensity score
Prior to intensity scoring, the scaling factor \(\sigma\) is calculated by comparing the intensities of the measured \(i\) and calculated \(j\) intensities for all peaks \(k\) within the matching threshold (see above). This scaling factor is calculated by dividing the weighted sum of the measured intensity by the weighted sum of the theoretical intensities (equation 4).
\begin{equation} \sigma = \frac{\sum\limits_{}^k intensity_{ik} r_{k} }{\sum\limits_{}^k intensity_{jk} r_{k}} \end{equation}Using this scaling factor, which is equal to the abundance of the measured molecule, one can calculate \(\delta^{intensity}_{ijk}\), which is the relative intensity error between measured and theoretical intensity for each peak \(k\) (equation 5).
\begin{equation} \delta^{intensity}_{ijk} = \frac{ \left|intensity_{ik} - \sigma intensity_{jk}\right|}{\sigma intensity_{jk}} \end{equation}The intensity score of peak \(k\) is then defined (equation 6).
\begin{equation} s^{intensity}_{ijk} = 1 - (\frac{\delta^{intensity}_{ijk}}{1 - r_{k} + \epsilon }) \end{equation}In analogy to the m/z score (\(s^{mz}_{ijk}\)), the denominator defines the range in which the peak based intensity score decreases from 1 to 0. However, in contrast to the m/z score, the intensity error has to be weighted by the abundance of each peak (1 - \(r_{k}\) ) as more abundant peaks can be measured more accurately than smaller peaks. Additionally, we introduced ϵ (pyQms parameter “REL_I_RANGE”, default 0.2), which represents the most conservative relative error applied to the most precisely measured peak (\(r_{k}\) = 1). Thus, the overall relative error (denominator) will increase with lower peaks The total intensity score \(S^{intensity}\) is the weighted sum of all similarity scores \(k\) in analogy to the \(S^{mz}\) score:
\begin{equation} S^{intensity} = \frac{\sum\limits_{}^k s^{intensity}_{ijk} r_{k} }{\sum\limits_{}^k r_{k}} \end{equation}The combined final score: mScore The final score is termed mScore and is a sum of \(S^{mz}\) and \(S^{intensity}\). However, because some machines can measure m/z much more accurately then intensities, we introduced \(\xi\) to allow for flexibilities depending on the type of mass spectrometer used. \(\xi\) (the pyQms parameter “MZ_SCORE_PERCENTILE”, default 0.4) is the fraction the \(S^{mz}\) score is weighted into the sum. Thus, the final mScore is defined as:
\begin{equation} mScore = \xi S^{mz} + (1 - \xi) S^{intensity} \end{equation}- matched_peaks (list of tuples) –